What is the finding
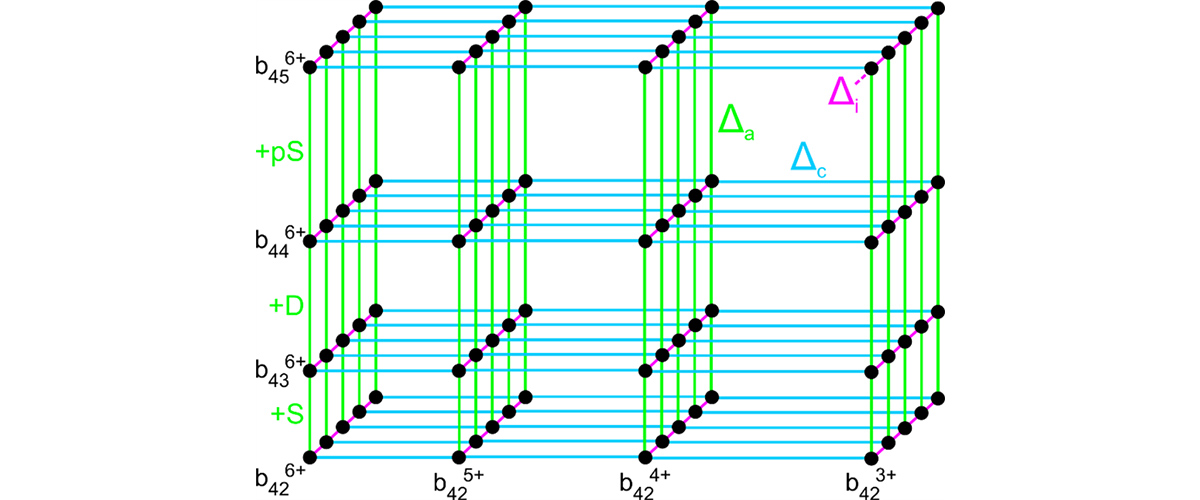
Scientists developed a new way to analyze large protein molecules by using patterns that naturally appear in mass spectrometry data, rather than relying on assumptions about what the protein should look like. By mathematically transforming the data, the method reveals consistent relationships that allow proteins to be interpreted more accurately, even when signals are crowded or distorted.
Why is this important?
Proteins are essential to life, and small changes in their structure can strongly influence how cells function, how diseases develop, and how treatments work. Accurately analyzing intact proteins is difficult because their experimental signals are often complex and overlapping, and traditional approaches can miss details or introduce errors when data do not match expectations.
This new method improves reliability by organizing experimental data into clear, consistent patterns that do not depend on prior knowledge of the protein. In addition to improving accuracy today, this structured representation creates a foundation for future artificial‑intelligence tools that could learn directly from experimental measurements to automatically recognize protein features and variations, ultimately helping to accelerate discoveries on disease biomarkers, drug targets, and deeper understanding of how proteins function.
Who did the research?
Lissa C. Anderson1,2, Nathan K. Kaiser1, Krishna Saketh Kamadana3, Xian Mallory3
1National High Magnetic Field Laboratory, FSU; 2Department of Chemistry and Biochemistry, FSU; 3Department of Computer Science, FSU
Why did they need the MagLab?
The National High Magnetic Field Laboratory provides access to one of the world’s highest‑field mass spectrometers, which offers exceptional resolution and signal stability. These capabilities are essential for detecting the subtle data patterns that make this new analysis possible and for generating the high‑quality, information‑rich data needed to support future AI‑driven interpretation.
Details for scientists
- View or download the expert-level Science Highlight, Connectivity‑Driven Sequencing of Intact Proteins
- Read the full-length publication, Mass-Invariant Natural Log-Transformed Mass Spectra Enable Internal Calibration and De Novo Sequencing of Intact Proteins, in Analytical Chemistry
Funding
This research was funded by the following grants: K. M. Amm (NSF DMR-2128556); Xian Mallory (NSF CCF‑2523717)
For more information, contact Kristina Hakansson.